识病寻源|CGD
·化脓性汗腺炎限制了患者工作和社交的能力,显著降低了患者的生活质量,并导致其收入下降、社会地位降低等等。许多患者同时具有抑郁和焦虑。一方面炎症可能导致心理疾病的进展,另一方面也有社会对皮肤病的污名化导致患者主动或被动的疏离。
2001年,三十多岁,事业初成的纽约电视新闻制片人米格尔桑乔(Miguel Sancho)和报纸记者费莉西亚(Felicia)在报道911事件中相知相识,他们都来自于中产家庭,有共同的热爱和趣味,情投意合,很快走到了一起。他们在纽约49街买下了一套公寓并组建了一个小家庭。在2008年和2012年他们先后孕育了两个孩子Lydia和Sebastian,准备将一个纽约经典爱情故事进阶为儿女双全的美式理想生活。然而,出乎桑乔的意料,儿女双全的生活没有想象中那么顺利。弟弟Sebastian出生数月,就频发高烧,常规退烧药物无效,且肛门附近反复出现浅红色的肿块。医生使用了大剂量抗生素甚至通过手术切除肿块来控制病情,然而Sebastian总会在一段时间后复发感染。就在反复就诊仍无法找到病因时,一位免疫学家给出了令桑乔震惊的诊断。
在询问病史、家庭环境之后,免疫学家做了严格的血液学检查。发现Sebastian体内一种主要对抗细菌和真菌的白细胞,中性粒细胞,不能正常地杀灭病原体。Sebastian的中性粒细胞数量足够,也很活跃,但在吞噬病原体后,病原体没有被杀死。这会导致身体产生更多的免疫积聚在感染部位,形成结节,这些结节被称作肉芽肿(granuloma)。这样的患者一生中会因为各类感染反复出现肉芽肿。免疫学家告诉桑乔,他的孩子得了一种罕见的免疫缺陷病,慢性肉芽肿(chronic granulomatous disease,CGD),发病率仅为25万分之一,而患者的平均预期寿命仅有30岁。
桑乔的家庭生活随着疾病的到来发生了深刻改变。为了创造一个尽可能没有病原体的环境,他和家人开始严格消毒,尽量减少与有菌环境的接触,保持“社交距离”。经基因检测后发现,Sebastian的X染色体上有CYBB基因的突变,从而解释了CGD的发生。他的妻子陷入了抑郁和愧疚,因为她觉得是她把有问题的X染色体传给了儿子。而桑乔的情绪也逐渐崩溃,开始酗酒、吸大麻麻痹自己。孩子的疾病侵蚀着父母的心理健康,桑乔负责的电视节目因此发生了严重播放事故。更严重的是,在愧疚、暴躁、抑郁等情绪的影响下,夫妻的感情纽带难以维系,婚姻濒临破裂。后来桑乔将这段经历写成了一本非虚构作品《你无法承受之重(More than you can handle)》,以记录家庭在遭受罕见遗传病中的艰难历程。

米格尔桑乔根据亲身经历创作的非虚构作品,2021年出版。 来源:https://www.amazon.com/More-Than-You-Can-Handle/dp/0593085914
一个重要的杀菌机器
1954年,在美国儿科学会的一次会议上,后来荣获拉斯克奖的著名免疫学家Robert Good等人首次报告了一组男性患儿,患有致命的反复感染。随后的几年里,医生们归纳特征,将其认定为一种疾病实体,称为“儿童致命肉芽肿”,因为很少有患儿能活过十岁 [1]。即使到上世纪80年代,仍有半数患儿在10岁前死亡 [2]。这是一种罕见的原发性免疫缺陷病,其特征是反复发生危及生命的细菌、真菌感染,并伴有组织肉芽肿形成。1967年,Robert Good等科学家们发现,这类患者的中性粒细胞与健康人不同,无法杀死细菌 [3];患者的白细胞在接触病原体时,也缺乏典型的氧耗量突然增加的“呼吸爆发”现象 [4] [5]。这一发现揭开了该病的病因学面纱。

Robert A. Good, M.D., Ph.D.,明尼苏达大学医学院教授,1970年拉斯克奖获得者,发现胸腺在免疫生物学中的关键作用,首次成功实施同种异体骨髓移植用于治疗严重免疫缺陷病。被称为现代免疫学的奠基人之一。 来源:https://laskerfoundation.org/
如今我们知道,这种疾病就是CGD,有不同的遗传模式,但都是由于烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶复合物出错导致的 [6]。NADPH氧化酶复合物由五个亚基及一个辅因子组合而成,主要表达在各类吞噬性白细胞上,包括中性粒细胞、嗜酸性粒细胞、单核细胞和巨噬细胞等。以中性粒细胞为代表,这些行使吞噬作用的免疫细胞吞入病原体后,可形成包裹病原体的囊泡。NADPH氧化酶复合物在这个囊泡上组装,随后通过消耗细胞质里的NADPH和囊泡内的氧气,催化产生大量过氧化氢等活性氧,进而转变为次氯酸等强效杀菌剂。这种依赖于氧气的杀菌作用是免疫细胞的一类重要杀菌方式,相当于在吞噬细胞内构造一个小环境围困病菌,然后释放强氧化剂杀灭它们。NADPH氧化酶复合物则是强氧化剂的制造机。
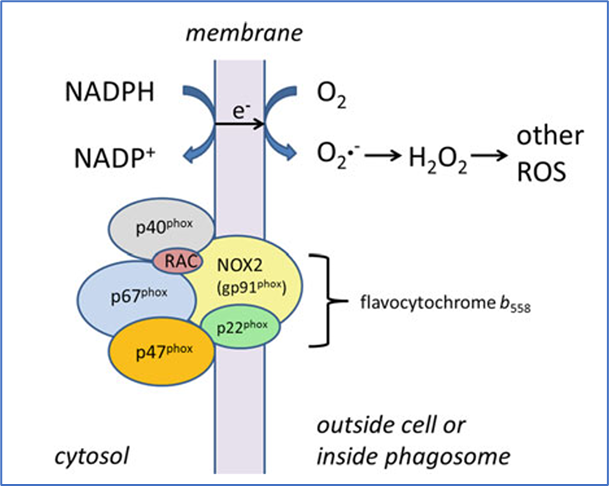
在吞噬囊泡上组装的NADPH氧化酶复合物。来源:[7]
这个机器的每个零件都对依氧杀菌过程至关重要。因此不同亚基的突变,不管是降低了活性氧的生产率还是导致机器完全失效,都可导致CGD。最常见的CGD是由编码一个关键亚基的CYBB基因突变所导致的。这个基因位于X染色体上,所以这类CGD属于X染色体隐性遗传病。若母亲的一条X染色体携带该致病突变,虽然自己有健康的表型,但可能将这一突变传给仅有一条X染色体的儿子,并导致其显性发病。这类X连锁型CGD占所有CGD的三分之二,且患者大多数是男性。这个机器的其他亚基的基因位于常染色体上,因此这部分常染色体型CGD患者男女比例相同。
诊断CGD,需结合临床症状与实验室检测。实验室检测除了开展针对NADPH氧化酶复合物的各个基因的检测之外,还需要对患者中性粒细胞开展测试,包括检测其活性氧产生、氧耗、NADPH氧化酶的各亚基表达情况、更进一步地还有中性粒细胞杀灭某些病原的能力等等。如果患者表现出残留的NADPH氧化酶活性,则预示着较轻的临床症状。残存的活性氧产生能力越高,患者的死亡率就越低。这些测试为提供遗传咨询、选择合适疗法、预测疾病进展奠定了基础 [7]。
无处躲避
CGD患者常常在婴儿期就开始持续感染,常遇到的细菌种类包括金黄色葡萄球菌、铜绿假单胞菌、诺卡氏菌等等。许多对免疫健全者通常无害的细菌,在CGD患者中却可能造成严重感染。例如分枝杆菌对CGD患者可构成威胁,这些患者在接种卡介苗(一种减毒牛分枝杆菌菌株)后,可能发展为严重的局部或全身性疾病。因此,CGD患者应接受常规儿童疫苗接种,但需排除卡介苗。有几种细菌几乎只有CGD患者才会感染,例如存在于温咸水中的弗朗西斯菌等,一旦发现应高度怀疑CGD。真菌感染对CGD患者也十分危险。很多真菌如烟曲霉菌等分布广泛,可以从草坪、树木、动物饲料中接触到,这些常见的霉菌或酵母菌一旦感染,可能会引起真菌性肺炎,出现弥漫性肺结节。
在CGD中,由于病原体无法被杀灭,促炎信号长期失调,炎症成为了持续的慢性状态,导致感染处免疫细胞聚集,形成一个个充满免疫细胞的肿块。肿块中存在着吞噬细胞、辅助性T细胞和B细胞之间复杂的相互作用,还可促进纤维化导致器官损伤,甚至挤占健康组织,导致继发疾病。这些肉芽肿根据感染部位不同,主要发生在肺部、胃肠道和泌尿生殖道。半数CGD患者同时患有炎症性肠病,其中结直肠区域受累最严重,因此常见患儿肛周肿块。CGD显著降低患者生活质量。虽然患者终身用药,仍难以避免反复出现的感染。由于缺乏社交,成年患者普遍存在焦虑等症状,儿童也面临情绪困扰。患者的学习、工作也被严重妨碍,很多患者处于停学、失业状态。
从治疗到治愈
对于急性感染的治疗,因感染早期症状不清晰,需检查炎症指标(如血沉率和C反应蛋白等),配合影像学检查和微生物学诊断。有时也需要做组织活检。治疗时需要使用抗生素和抗真菌药物,如伏立康唑等。但患者感染风险终生存在,不能只依赖感染后的治疗,而需要进行预防。除了创造无菌环境外,还需终身使用预防性抗生素和抗真菌疗法,如联用甲氧苄啶-磺胺甲噁唑(TMP/SMX,一种抗生素复合制剂)和伊曲康唑(广谱抗真菌药) [8]。CGD的大部分治疗旨在控制感染及炎症并发症,目前已经形成相对规范的药物方案。特定情况下,也可合并使用干扰素或激素治疗。有时,需要使用手术引流或者手术切除受感染部位的脓肿,才能帮助患者恢复。但总体而言,这些疗法还是“治标”的方案,疾病的本质--杀菌功能缺陷--仍未改变。
治愈CGD是科学家和医生们的终极目标,目前临床上可用的治愈方法是造血干细胞移植,用新的造血干细胞置换当前全身血液细胞,特别是无法正常工作的中性粒细胞。由于CGD患儿一般比较年幼,骨髓移植后的生存率较高。然而临床上骨髓移植面临两大难题,一是特定风险,如移植前要对患者骨髓预处理,抑制患者的免疫系统;而移植后可能会出现移植物抗宿主病,即移植物构成的免疫系统错误地攻击患者的组织器官。这种情况下需要免疫抑制药物来减缓攻击。另一个问题是供体不足,由于造血干细胞移植需要HLA配型相合,即使是亲兄弟姐妹也不一定能够达标。所以患者常常难以找到合适的供体。于是,针对CGD患者的基因治疗就提上了日程。
CGD的基因治疗有三种策略。第一,基于慢病毒载体,分离出患者自体造血干细胞后利用病毒将正常基因导入,令其表达。在一项对X连锁型CGD的临床试验中,治疗后患者的中性粒细胞具有稳定的载体拷贝数,12个月随访时,9名患者中有6名有16%-46%的氧化酶阳性中性粒细胞。基于这一积极结果,美国食品药品监督管理局(FDA)于2020年1月授予该基因治疗OTL-102罕见病药物认定,用于治疗X连锁型CGD。但这种将一份额外的基因整合进入细胞的基因治疗仍有不可避免的插入致突变风险。
第二种方法是利用斩获诺奖的CRISPR-Cas9技术,对基因上出问题的位点开展基因编辑。该系统通过引导RNA的定向作用,使CRISPR/Cas9核酸酶能够在目标位点诱导DNA双链断裂,从而引入修复。该方法的主要风险在于CRISPR具有一定几率脱靶,同时基因编辑效率尚不足以达到有效治疗的水平。
第三种方法是一类新兴的、致力于减少基因组错误的基因编辑方式,prime编辑。该技术由获得2025年科学突破奖的哈佛大学教授David Liu(刘如谦)实验室在2019年基于CRISPR-Cas9技术开发。相比于传统的CRISPR-Cas9技术,prime编辑有特殊优势,即它不需要在目标DNA上做双链切割。科学家们改造了Cas9,这种Cas9只切割其中一条互补DNA链,从而打开一个可以插入新序列的空缺,进而在引导RNA的帮助下合成新链,实现“以新换旧”。这个策略导致脱靶几率大大降低,增加了安全性。在麻省理工学院Robert Langer教授等著名科学家的共同努力下,新版本的prime编辑器经过不断修正,大大增加了基因编辑的精准性 [9]。
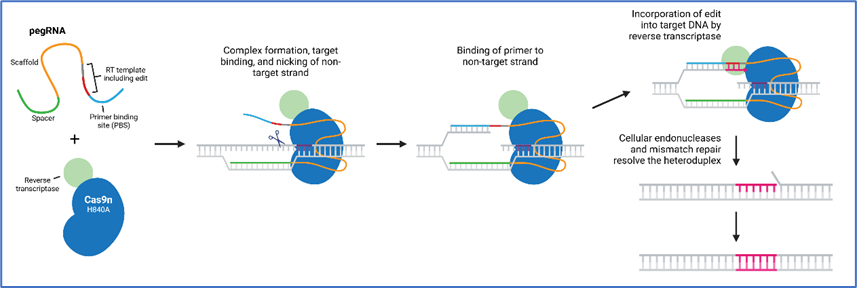
Prime editing的工作原理。来源:https://blog.addgene.org/prime-editing-crisp-cas-reverse-transcriptase
2024年4月29日,生物技术公司Prime Medicine宣布(https://primemedicine.com/),其用于治疗CGD的新药临床试验申请已获美国FDA批准。该疗法PM359通过体外prime编辑患者自身的造血干细胞,纠正关键基因的一个常见突变。2025年5月19日,《自然》杂志报道了令人振奋的初步结果,一位18岁的CGD青年患者接受了prime编辑疗法 [10]。在治疗后第15天,58%的中性粒细胞通过功能测试;到第30天,这一比例达到66%——远高于临床获益阈值(约20%)。在治疗一个月后,尚未报告与PM359相关的严重不良事件。这是prime编辑技术首次应用于患者治疗。目前公司已有计划在多种遗传病上推进这一疗法。也许在不远的未来,CGD患者将不再受制于抗感染治疗及艰辛的骨髓移植程序,可以迅速恢复免疫力并回到生活轨道。
回到桑乔的故事,在咨询美国多家医疗机构后,Sebastian在杜克大学儿科血液和骨髓移植项目中注册,并幸运地找到了配型合适的供体。2016年,在经历了令人揪心的的移植程序后,小Sebastian已经重建了免疫系统,获得了有功能的中性粒细胞。随着疾病的治愈,家庭也被治愈,桑乔现在是一个成功的电视制作人,而母亲费利西亚成立了美国CGD协会(https://cgdaa.org/),专为CGD患者和携带者提供服务。

Sebastian与母亲、姐姐和父亲的合影。来源:https://primaryimmune.org/resources/news-articles/book-chronicles-struggle-cgd-diagnosis
从上世纪五十年代"致命肉芽肿"的绝望诊断,到今天最新的基因编辑技术带来的治愈曙光,CGD的治疗历程跨越了基础医学、遗传学、免疫学、传染病学、移植和基因治疗等多个领域,见证了现代医学的进步。当代的CGD治疗始于严格的感染防控,历经造血干细胞移植,正迈向基因编辑的创新时代:通过精准修复错位的遗传指令,重建免疫细胞里能力非凡的“杀菌机器”。让曾经的"不治之症"逐步变为可控、可治、乃至可治愈的疾病。每一次技术迭代,带来的是更稳定的免疫重建,更少的住院,更多离开无菌环境回归日常生活的机会。而医学的意义,也就在免疫缺陷患者重返日常的脚步里。
(作者杨云龙,系复旦大学基础医学院细胞与遗传医学系教授、系主任,在中国细胞生物学会医学细胞生物学分会、中国优生科学协会基因诊断与精准医学分会兼职。作者孙维泽,系复旦大学华山医学院临床五年制本科生。疾病不断地改变着每个人的人生轨迹。但除了医生与医学研究者,人们很少有机会了解各式各样的疾病。“识病寻源”专栏将以一文一病的形式,介绍对疾病的认识进程,疾病的病因及其治疗。跟随医学科学的进步,理解现代医学。)
参考文献
1. Berendes, H., R.A. Bridges, and R.A. Good, A fatal granulomatosus of childhood: the clinical study of a new syndrome. Minn Med, 1957. 40(5): p. 309-12.
2. Mouy, R., et al., Incidence, severity, and prevention of infections in chronic granulomatous disease. J Pediatr, 1989. 114(4 Pt 1): p. 555-60.
3. Quie, P.G., et al., In vitro bactericidal capacity of human polymorphonuclear leukocytes: diminished activity in chronic granulomatous disease of childhood. J Clin Invest, 1967. 46(4): p. 668-79.
4. Baehner, R.L. and D.G. Nathan, Leukocyte oxidase: defective activity in chronic granulomatous disease. Science, 1967. 155(3764): p. 835-6.
5. Holmes, B., A.R. Page, and R.A. Good, Studies of the metabolic activity of leukocytes from patients with a genetic abnormality of phagocytic function. J Clin Invest, 1967. 46(9): p. 1422-32.
6. Yu, H.H., Y.H. Yang, and B.L. Chiang, Chronic Granulomatous Disease: a Comprehensive Review. Clin Rev Allergy Immunol, 2021. 61(2): p. 101-113.
7. Roos, D., Chronic Granulomatous Disease. Methods Mol Biol, 2019. 1982: p. 531-542.
8. Zerbe, C.S. and S.M. Holland, Functional neutrophil disorders: Chronic granulomatous disease and beyond. Immunol Rev, 2024. 322(1): p. 71-80.
9. Chauhan, V.P., P.A. Sharp, and R. Langer, Engineered prime editors with minimal genomic errors. Nature, 2025.
10. Ledford, H., World first: ultra-powerful CRISPR treatment trialled in a person. Nature, 2025. 641(8065): p. 1083.
相关文章
内外“双卷”、创新惰性,独角兽增速下滑折射出的产业发展困境

特朗普赦免币安创始人赵长鹏

美国债务首次突破38万亿美元,政府停摆和“大而美”法案加剧危机
但斌最新美股持仓曝光:首度建仓阿里、博通,英伟达仍为第一大重仓股

浙江前三季度GDP为68495亿元,同比增5.7%

高连奎评《货币、金融、现实与道德》|债务奴役:利息沦为现代化的贡品?

全球前五,上海迪士尼2024年客流量突破1470万人次

一周文化讲座|闯入诺奖得主拉斯洛的文学宇宙

《再见爱人5》第二期今日播出

圆桌|美国B-1B轰炸机逼近委内瑞拉,拉美如何成为特朗普的后院秀场

真实、娱乐与幻想:东方“美人鱼”的西方之旅

包茂红|高山之巅:印加帝国的兴亡与环境
圆桌丨美国B-1B轰炸机逼近委内瑞拉,拉美如何成为特朗普的后院秀场

广西前三季度GDP为21487.03亿元,同比增5.3%

董岚已任湖南省高院党组副书记、副院长

韩国公布梨泰院踩踏事件监察结果:计划惩戒62人

桂林十如调研:纺织行业绿色低碳转型,理念如何变为成效?

中共中央新闻发布会即将召开,中外媒体关注这些问题
韩国政府公布梨泰院踩踏事件监察结果:计划惩戒62人
